


HapMap五周年回顧
作者簡介:曾長青,中國科學院北京基因組所研究員,博士生導師。CUSBEA獎學金、杰出青年基金、首批新世紀百千萬人才工程國家級人選獲得者。作為“十五”重大攻關項目課題組長、國際HapMap計劃Steering Committee Member和 “中華單體型圖協作組”召集人,負責HapMap“中國卷”的實施。主要從事疾病相關基因定位、群體遺傳學和基因組多態研究,部分成果在Nature,Nature Genetics, PNAS等雜志發表。
今年10月,是人類基因組國際HapMap計劃啟動8周年和這一重大國際合作的主要任務完成5周年。幾年間,全球范圍的基因組特別是人類基因組的研究,已經成為生命科學的最前沿學科,HapMap也成為應用最為廣泛、深入并不斷完善更新的人類最大數據庫。本文回顧性介紹HapMap計劃及其近年來在基因組學和人類健康領域的重要作用。
1、人類基因組的HapMap和國際HapMap計劃
(1)何謂HapMap
HapMap是Haplotype Map 的簡稱,Haplo意為單一,在基因組中專指來自父母的一對染色體中的一條。Haplotype就是單條染色體中的一段,譯作單體型(有人譯作單倍型),是描述遺傳差異的一種主要方式。DNA作為遺傳物質,不但編碼了物種間的差異,物種內不同個體之間的差異也含在其中,均表現為基因組之間的DNA序列差異,也就是基因組的多態性上。
DNA由四種核苷酸單個連接而成,基因組最常見的多態就是單核苷酸多態(single nucleotide polymorphism),簡寫為SNP或SNPs(復數),指在群體中染色體的某一位點上由不同的核苷酸構成(圖1)。
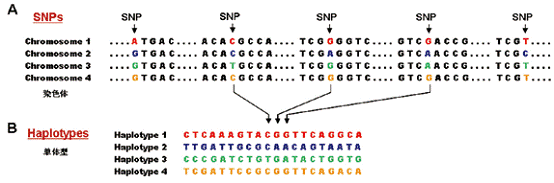
圖1 單核苷酸多態位點SNP和單體型。A:在來自4個個體的一段6kb序列上,大部分核苷酸相同但有5處顯示不同(彩色)即SNPs。B:這段6kb區域的20個臨近SNPs組成單體型(Haplotypes)。摘自Nature. 426:189-193。
目前發現的人類基因組中的SNP位點已經超過一千萬。在人群中,染色體上每一二百個核苷酸就有一個SNP位點。單體型描述的是一段單條染色體上的序列差異,就是由SNP位點的順序排列組成。因此也可以說單體型是分別來自父母的單條染色體上SNP的分布和傳遞模式。根據鄰近SNP的連鎖特性(即連鎖不平衡),單體型上的多個SNPs還可以由少數幾個tag即標簽SNP代表。Haplotype Map是單體型圖譜,就是全基因組上所有DNA序列的SNP分布和人群頻率、標簽SNPs、連鎖性質與規律等。
(2)HapMap的內容
HapMap的內容是一個巨大的“人類遺傳用表”。遺傳學研究的本質在于解析變異及其傳遞規律。對于人類基因組中的千百萬個SNPs來說,盡管很多SNPs對于表現型可能并沒有貢獻,個體的性狀差異,也就是任意兩個人之間的遺傳差別,就蘊藏在這些SNPs和單體型之中。如果說人類基因組測序計劃打開了我們自身的遺傳密碼這本天書,那么地球上每個人的天書都是一種版本,HapMap揭示的就是不同天書版本之間的差別與規律。從個體的基因組測序到全基因組SNPs在人群中的分布和單體型的構建,HapMap計劃標志著群體基因組學研究的開始,旨在通過對于海量SNPs的人群檢測及其計算分析,構建全人類的遺傳用表,從而研究者可以從中查到基因組中各種位置的SNP以及在主要人群中的組成、頻率和單體型與標簽SNP情況。
(3)HapMap的重要價值
HapMap的重要價值在于揭示復雜性疾病的遺傳因素。復雜性疾病是相對于單基因病(如血友病等)而言,也是常見疾病,如高血壓、腫瘤、精神性疾病、糖尿病等。其發生往往是遺傳和環境因素共同作用的結果,遺傳因素涉及多個基因和分子通路,而不表現為單基因的孟德爾遺傳。因此罹患這類疾病的易感性,包括對不同環境或藥物的敏感性等,都隱匿在多個基因的微效變異即SNP和單體型及其組合中,并且有很大的個體差異。因此,揭示這類疾病的遺傳模式需要大量的群體樣本和發現與檢測極多的SNPs進行關聯分析,這在HapMap構建之前幾乎是不可能的。國際單體型圖計劃(TheInternational HapMap Project)的主要目的就是構建不同人群的高密度SNP圖譜,通過分析計算確立單體型及其中SNPs的連鎖性質和標簽SNPs,從而使研究人員可以根據這一巨大的遺傳圖表和所揭示的人類群體的分子遺傳機制,為發現復雜性疾病的相關易感基因確定研究方案和選擇需要進行分析的標簽SNPs。
(4)HapMap計劃的啟動和主要任務的完成
國際單體型圖計劃經過3年的籌劃,于2002年10月29日在華盛頓召開由加拿大、中國、日本、尼日利亞、英國和美國6個參加國代表出席的第一次戰略會議。會后協作組(Consortium)召開新聞發布會向全球宣布這一計劃的正式啟動。HapMap計劃參加國中加、中、日、英和美國分別承擔全基因組10%,10%,25%,24%和31%的SNP分型任務。尼日利亞提供非裔樣品,中國和日本共同提供亞裔樣品,美國提供了歐裔樣品。第一次戰略會議上決定了HapMap兩階段的戰略。第一階段是針對非、亞、歐裔的270個DNA樣品在全基因組范圍以平均每5 000個核苷酸(5 kb)一個SNP的密度進行大規模SNP分型鑒定,構建5 kb單體型圖。第二階段是將HapMap的分型密度增至2kb左右。HapMap計劃的運作由各國代表組成的Steering Committee及其下屬的若干工作小組負責執行實施。
3年后的2005年10月26日,由國際協作組總負責人,現任美國NIH負責人Francis Collins向全世界宣布了一個擁有數億數據的人類基因組單體型圖的成功構建,以及一個更精細的遺傳圖譜即將完成(圖2)。

圖2 2005年10月26日美國鹽湖城HapMap新聞發布會場,國際協作組負責人,時任美國國立人類基因組研究所所長FrancisCollins代表所有參加國和國際協作組宣布人類基因組單體型圖的圓滿完成。
各中心的巨大努力使分型密度比預計提高了近40%,共針對一百多萬SNPs構建了密度約3.6kb的數據庫和HapMap。美國衛生和公共服務部部長Mike Leavitt在會上評價和描述了HapMap計劃的意義和前景。新聞發布會上還發行了載有HapMap及其長文“人類基因組的單體型圖譜”的Nature雜志。HapMap這一任務的完成,也標志著這個巨大項目的“中國卷”的完成。中國科學家為這一計劃做出了10%的貢獻。
(5)HapMap計劃的后期工作和進展
HapMap計劃的第一階段任務完成后,國際協作組委托Perlegen Sciences 完成第二階段擴大SNP分型密度的任務。2007年10月18日,國際協作組在Nature上發表了根據第二階段數據構建的人類基因組的第二代HapMap。至HapMap 二期共發現了超過一千萬的人類基因組的SNPs,完成了約310萬SNPs(≥5%)在270個樣品中的分型反應。這些SNPs約占預測的遺傳變異的25%~35%,并使第二代HapMap的分辨率達到平均不到1kb一個SNP,比預定計劃超過100%,準確度達到99.8%。
為使HapMap具有更大參考性,在前兩個階段近10億SNP數據的基礎上,一個新的人群分析HapMap 3啟動并于2010年9月在Nature上發表了新的海量數據。與前兩期內容不同的是,HapMap 3旨在大量擴充人群樣本和發現低頻率SNPs。共160萬的常見SNPs在來自全球11個人群的1 184個體中進行了分型反應,使HapMap具有更廣泛的代表性。同時,還在其中692樣品中進行了1Mb區域(10kb×10)的重測序,以發現新的低頻率SNPs。顯然,隨著更多數據的產生,人類基因組的HapMap將不斷更新使之涵蓋更多人群特異數據和具有更為精細的分辨率。
2、重新了解的基因和染色體——HapMap對于人類基因組結構的重大貢獻
(1)基因含義的修訂
早在2005年之初,HapMap尚未完成之時,Science就預測這一計劃的完成將是生命科學取得的最重大進展之一;至當年底HapMap又被兩院院士評為世界10大科技進展中的第5位。的確,HapMap計劃的結果與應用及其由此引出的更多基因組規模的深入研究帶來了遺傳學和基因組學的全面知識更新,甚至包括現代分子生物學概念中的基因與染色體的結構。作為HapMap的衛星項目開始的ENCODE 計劃(Encyclopedia Of DNAElements,DNA元件百科全書)于2007年宣告完成。這一擴展到涉及11個國家80家機構參加的研究通過整合計算測序數據和實驗分析,鑒定出人類基因組中所有的功能組分,包括編碼基因、非編碼基因、調控區域、染色體結構維持和調節等所有類型DNA元件的分布和組織方式。合作組同時在Nature和Genome Research上發表了29篇論文,詳盡描述了在所分析的1%人類基因組區域內最為完整的生理功能元件,還對很多傳統的分子生物學定義做出顛覆性擴展或修訂。例如,microRNA,非編碼RNA等的普遍存在使“一個基因一個酶”,“中心法則”等都不再是生命科學的金科玉律。這些革命性進展為進一步認識基因組的功能藍圖開辟了道路,對疾病的遺傳研究產生了重大影響。
(2)全新的結構差異和拷貝數變異概念
雖然HapMap計劃的目標是對于單核苷酸多態位點的模式分析,一個未曾預見的重大成果是通過對于單體型的分析所發現的基因組中同樣廣泛存在的結構差異(structural variation,SV),包括DNA序列的插入、刪除、倒位、易位等。這些結構差異大小很不均一,可在數十個至數百萬核苷酸之間。HapMap 揭示出結構變異是基因組中一種常見但由于經典檢測技術的缺陷而知之甚少的遺傳多態性。由于很多結構多態可在HapMap這一高密度差異圖譜上留下“印記”,近年來隨著對HapMap的分析利用使人類對于自身染色體的結構多態性產生了全新的認識。僅2006年就有3個小組從不同角度構建了人類基因組的缺失圖譜。隨著對DNA片段的插入和刪除愈來愈多的發現,拷貝數變異(copy numbervariation, CNN)成為描述這類遺傳差異的最新名詞。這類新發現的遺傳變異覆蓋了人類基因組大約20%的區域,估計10%~20%的可“調節”基因活性的遺傳變異是CNV。同SNP一樣, CNV可導致先天性疾病并與多種復雜性疾病的發生有關。HapMap的完成不但導致了人類基因組拷貝數目多態性計劃(The CNV Project)的啟動,也為高通量篩查CNV 提供了全新的研究策略和實驗手段。需要指出的是,拷貝數變異多發生在重復序列,目前對于“斷點”位置的精細確定極具技術挑戰性。因此一些研究曾導致CNV在基因組中的覆蓋度達40%~60%的過高估計。準確定位CNV有待于更先進手段如單分子測序等的發展。
3、HapMap計劃對于基因組科學和系統生物學的極大推動
如果說人類基因組計劃奠定了基因組學的基礎,HapMap計劃則開啟了群體基因組學的時代,并且在很大程度上參與催生了系統生物學的發展。HapMap提供了全基因組SNPs 的群體分布圖譜并揭示了人群內的遺傳結構,不但構建了“群體基因組學(populationgenomics)” 基礎還建立了一種新的研究策略。這些使自然選擇和人群演化成為近年來基因組學研究的又一熱點。基于此,近年來還獲得了若干人類陽性自然選擇圖譜和數據庫。尤其值得一提的是,2010年我國多個小組通過獨立或合作研究,數月間先后在Science,PNAS,Mol. Biol. Evol.刊登多篇文章闡述藏族對于高原低氧適應性的分析結果,所有研究均采用HapMap的漢族數據作為對照進行比較。這些成果不但為深入的群體遺傳學研究和疾病的遺傳因素及基因功能分析提供了新的視野,還大大加深了人類對于環境的適應過程和人群表型分化的認識。
HapMap計劃不但帶來更多全基因組規模的大型研究,還大大推動了對多個物種的系統基因組學研究。如上述ENCODE 計劃完成之后,一個更大規模的多物種ENCODE項目又開展起來,以解析ENCODE保守區域在不同物種中的細節,進一步了解物種演化過程中的基因組系統變化。利用HapMap數據進行的基因表達研究,還為分子通路和網絡研究奠定了數據基礎。正是這些研究的匯集和整合,促進了系統生物學的產生和深入開展。
4、應用遺傳用表解析疾病原因
人類遺傳用表HapMap已經廣泛用于疾病研究。HapMap計劃的立項目的是為復雜性疾病的基因定位研究提供基礎數據、研究策略和先進技術。在此之前,復雜性疾病的遺傳研究始終缺乏有效方案。主要原因在于這些疾病不但是遺傳和環境因素共同作用的結果,并且其遺傳因子涉及多個基因和多條基因產物互作的分子網絡或代謝通路。在這類疾病的遺傳因子中每個基因的變異對于疾病的貢獻并不顯著,但其某種未知整合卻成為個體罹患疾病的風險。同時,這些不同的易感基因或位點在疾病中的作用有很大的個體差異。因此,如果在群體規模將所有的SNPs進行篩查以確定疾病的相關基因,這在耗資上是無法做到的。HapMap計劃應用基因組學“單體型板塊”、“標簽SNP”等原理,一方面描述了個體差異位點的分布和傳遞規律,另一方面構建了以人群中遺傳差異的傳遞模式為主要內容的圖譜。其中無冗余的分型SNPs超過310萬,連同用于質控和低頻率SNP位點,整個HapMap計劃在270個樣品中進行了分型的SNPs達到630多萬,超過所估計存在的人類SNPs數量的一半。這些供全球免費下載的數十億份數據,為各種規模的病例-對照關聯分析提供了極為重要的工具和數據,使大規模的對于復雜性疾病的關聯分析成為可行的易感基因定位手段。對于較為傳統的使用家系樣品進行的單基因病連鎖分析,HapMap的海量數據使高密度SNPs成為比傳統的微衛星標記有更高的解析度和準確率的遺傳標記,并使連鎖與關聯分析的聯合使用成為更為有效的定位方法。
從HapMap計劃對外發放數據,就揭開了通過SNP分型進行病例-對照的關聯分析的序幕。HapMap的完成使候選基因或全基因組的病例-對照關聯分析在全球范圍得到普及,并且促進了幾個“超大型”的復雜性疾病遺傳因素分析計劃的立項和實施,包括GAIN(geneticAssociation Information Network,遺傳關聯信息網絡)、POPRES(POPulation REference Sample,群體參照樣本)、WTCCC(The Welcome Trust Case-Control Consortium,Welcome基金會病例對照協作組)和多個針對某一疾病如高血壓等的大型聯合研究。其共同特點是采用包含標簽SNPs的全基因組芯片,進行GWA(genome-wideassociation)研究,又稱GWAS(GWA studies),即不考慮先驗知識直接通過對大樣品量(上千份)進行整個基因組的關聯分析研究。例如,2007年11月WTCCC報告了對超過16000個樣本所進行的針對雙向情感障礙等7種常見復雜性疾病的GWAS結果,在克羅恩病(Crohn’s disease)等5個疾病中發現至少一個易感基因信號。GAIN 計劃則為注意力分散/多動癥等6種復雜性疾病的GWAS研究提供支持,尋找易感基因和新的診斷方法。HapMap作為人類遺傳用表已經廣泛用于疾病相關基因定位研究。
復雜性疾病相關基因定位研究是長期探索和緩慢積累的過程。然而,大量全球性的大規模GWAS研究亦逐漸暴露出許多問題和局限,包括部分大型研究沒有發現顯著信號,不同人群結果重復性差,無法解釋疾病相關信號的生物學意義,易感性對于疾病的真正貢獻缺乏評估和與指導臨床相脫節等。這些實際上顯示出揭示多基因常見疾病遺傳機制的復雜性和難度,也反映了一些初期對于使用現有方法一舉解決復雜性疾病遺傳機理的期待可能過于樂觀簡單。對于一種涉及多種分子通路的復雜性疾病來說,發現其所有的遺傳因素也許如同完成一幅復雜的拼圖(jigsaw),需要大量元件的積累和逐漸拼接。以現有基因組學知識考慮和綜合分析GWAS結果,人群的遺傳背景差異和部分易感性位點是較低頻SNP可能是GWAS信號不顯著和人群重復性差的重要原因。此外,樣品收集過程中臨床標準的取舍、分類、控制等方面的差異,也可能降低重復試驗的分辨率。對此,將千人基因組等項目發現的低頻率SNPs也整合至GWAS研究中,以及擴大樣品量或進行若干個類似GWAS的聯合分析即Meta analysis,將使發現陽性信號的可能性大為提高。最近,一個由超過100個研究中心參加的糖尿病相關國際協作組對總共8 000多個病人和近40 000對照進行薈萃分析,發現了12個與胰島素分泌相關的II型糖尿病的易感基因和位點。此外,隨著越來越多的遠距離調控因子及非編碼RNA的發現,非基因區的信號也將獲得越來越多的解釋。毋庸置疑,與基因組學及其先進技術的迅速更新相比,在HapMap基礎上以關聯分析為主的大規模復雜性疾病易感基因的定位研究無疑將是一個長期探索和緩慢積累才能最終全面整合結果的過程。
5、HapMap計劃對于中國基因組科學的重大推動
如同人類基因組計劃,HapMap計劃的完成是一個里程碑式的成就,從研究策略到分析與推算的多種手段和算法,均代表了最先進的科研成果和進展,具有極高應用性和創新性。HapMap“中國卷”為構建占人類基因組10%的3號、21號染色體和8號染色體短臂的單體型圖以及提供一半的亞洲樣品。主要內容于2002年納入國家“十五”科技攻關計劃,其中2.5%的任務由香港創新科技署和香港大學教育資助委員會聯合資助的香港小組完成。HapMap中國卷10%的任務量對于承擔團隊是極為嚴峻的考驗和挑戰。HapMap其他成員都是擁有高通量SNP分型體系和經驗豐富的一流中心,只有中國團隊從購買安裝設備開始,各方面從一開始就落后他人一年。特別是,北京課題組資金嚴重不足。雙重巨大壓力下團隊背水一戰,超負荷追趕,采取一系列措施提高反應能力和降低成本,節省一切可能經費,最終在精誠合作和相互幫助下,中國團隊完成了超過3 500萬的SNP分型反應,保質保量地完成了中國卷任務并成為最早完成補洞的國家。
從人類基因組計劃的1%到HapMap計劃的10%,中國同發達國家一起參加了揭示人類遺傳機制全貌的兩個宏偉計劃,也是其中唯一產生數據的發展中國家。1%項目的參與使我國在基因組學這一學科領域得到迅速起跑,HapMap10%的貢獻則標示著中國科學在基因組學這一新興學科中的飛躍發展。這一歷史過程不但顯示了我國在基因組學的巨大進步以及國民和國力對于這一人類最大公益項目的支持,還體現了中國科學家抓住歷史機遇、把握學科前沿進展,從落后領域沖天而起的拼搏與成功。中國的參與發展訓練了研究隊伍,并極大促進了基因組學在中國的開展。在中華民族的遺傳多態和重大疾病的基因組研究方面,多項重要研究成果和高水平論文每年都在遞增產生。我國在這一領域已經獲得大量世界領先成果并將以更大的生命力持續發展下去。
6、基因組科學帶動的先進技術手段的發展
基因組科學發展的一個重要特點就是與先進技術手段極其緊密的相互結合和促進,正是這種密切關系產生了科學與技術共同的迅猛發展。同早期測序技術落后的情況不同,多種比較成熟的SNP分型技術在HapMap計劃的實施之初已經發展起來,從而保證了HapMap計劃的3年圓滿完成。HapMap計劃不但有力推動了SNP技術的快速發展,還大大促進了多種基因組學技術的研發。特別是多種密度的全基因組SNP芯片為病例-對照研究提供的有效工具,幾乎被所有大型疾病相關研究采用。同時,在SNP雜交芯片基礎上,多種全基因組規模的其他分子檢測芯片陸續開發出來,包括定量檢測轉錄本表達、microRNA、DNA甲基化等檢測芯片,已在人和多種模式生物中獲得大量應用。通過基于SNP或微列陣的比較基因組雜交(comparative genomic hybridization,Array-CGH),則成為發現和研究拷貝數目差異的重要手段。
值得一提的是用于SNP分型的高通量芯片雜交技術還催生了應用微型“霰彈法”測序策略的二代測序技術的研發,近兩三年來以高通量為特點的新一代測序儀已經成熟并迅速占領了市場。無論在研究策略和手段,還是分析內容的種類和深度上,二代測序儀的普及都帶來了基因組學研究的革命性思路和進展。如群體基因組規模的測序項目千人基因組計劃的前期工作(Pilot project)已由二代測序手段完成,包括對兩個三體家系的深度測序(平均42X),159個HapMap樣品的低度測序(2-6X)和對697個體的部分外顯子測序。美國NIH啟動的腫瘤基因組計劃(The Cancer Genome Atlas,TCGA)目前正在以膠質細胞瘤、肺癌和卵巢癌為先導,通過二代測序進行腫瘤基因組的分析。此外,在SNP雜交芯片的基礎上與二代測序同時發展起來的還有通過分子探針進行基因組上目標序列捕獲的技術。根據研究需要的定制靶點探針和針對全基因組編碼部分的商品化外顯子探針將目標序列從整個基因組上特異性捕獲下來,與二代測序聯用,已經成為一種新的研究方法,極大促進了相關基因定位、醫學重測序等復雜性疾病研究。
7、公益事業的光輝典范
作為重大國際性公益事業,HapMap的成功合作和完成,再一次打破基因專利對于人類健康事業的挑戰。自人類基因組測序的工作框架圖發表,研究SNP和基因組多態性便成為新的研究熱點。特別是,人類基因組的SNP圖譜是通向確定復雜性疾病易感基因與位點的鑰匙,導致又一次出現了多個私營集團因基因專利的巨大商機而多方位興起的全基因組規模研究熱潮,再次使人類基因組計劃所提出的數據共享原則受到嚴重挑戰。由此,國際協作組提出了人類基因組研究的第二個戰略任務,以多國合作形式共同構建單體型圖的國際HapMap計劃應運而生。為了與可能的基因或SNP專利趕超時間,協作組特別采取了根據項目進度隨時公布數據的策略。在HapMap網站上將所有階段性數據即時發出供全球研究人員無償使用(www.hapmap.org)。最終HapMap的所有數據,包括低頻和罕見SNPs、分型技術、算法與結果等,全部無償公布,供全球其他研究所用。
這些舉措使人們再一次解除了對大規模疾病相關基因和位點的專利的憂慮。如同測序計劃的全人類共享運作與私營企業試圖搶先基因專利的激烈競爭的一個更大規模的重演,最終受政府支持的公益項目以其無法超越的規模和速度迫使多個啟動更早的企業相關項目,特別是針對復雜性疾病的計劃放棄對于SNPs的專利嘗試。不止一個企業有償或無償地將數據納入到HapMap計劃中,甚至從競爭轉為重要參加者。最終一個高密度的HapMap和目前擁有數十億數據的人類基因組多態數據庫成為全人類共有的寶貴財富。
還在HapMap計劃醞釀之時,少數資深學者對于這一耗資甚大的計劃所構建的HapMap能否為后續的疾病相關基因研究提供重要數據懷有疑問并影響到一些我國學者。同時還存在是否“值得”耗資參加這樣的公益項目的疑問。對此,自2004年開始顯露的全球范圍對于HapMap結果的大量應用已經充分證明了HapMap數據對于遺傳多態和基因組研究的不可替代的重要意義,HapMap的奠基用對于后續開展的系列全基因組規模研究的巨大作用實際上大大超出預料。而中國作為參加國之一,目前在基因組學方面獲得的矚目成就亦早已證明益莫大焉。HapMap計劃已經將基因組學研究引向新的階段,對人類健康產生著深遠影響。一個全面揭示所有復雜性疾病的相關基因和人類遺傳機制的時代正在來臨。
本文發表于:《科學新聞》中國生物研究熱點論文特約稿——Vol. 5 No. 6 2010


